


Traditional Taxonomy and Modern Phylogenetics
Traditional taxonomy initially relied upon grouping taxa by morphological similarity. As the study of systematics became more and more sophisticated, new methods and characters, such as presence or absence of various chemicals or chemical pathways, were integrated into the view of how various groups and species of plants are related to one another.
Much more recently, the advent of DNA sequencing has revolutionized the field of systematics and how phylogenetic trees are constructed. This shift began in earnest in the mid-1990s, and one of the earliest genetic loci used in molecular phylogenetics of plants was rbcL, the gene that encodes the large subunit of the carbon-fixing protein Rubisco. As DNA sequencing technology has improved and become less and less expensive, computational speed and availability have allowed for large datasets of tens of thousands or even hundreds of thousands of molecular characters less prone to convergent evolution or incorrect interpretation of homology than morphological characters to be analyzed.
There are actually 3 different genomes within a plant cell: the nuclear genome where the majority of genes reside; the mitochondrial genome, which is a remnant of the bacterial genome that has become much reduced since the ancient endosymbiosis of an ATP-producing bacterium in the ancestor of all eukaryotes; and the chloroplast genome, the remnants of the cyanobacterial genome from the endosymbiotic event in the ancestor of Archaeplastida (Red Algae + Green Algae/Embryophytes). The nuclear genome is encoded on large, linear chromosomes while both mitochondrial and chloroplast genomes map to circular plasmids like their prokaryote precursors.
Traditional Taxonomy and Modern Phylogenetics:
Traditional taxonomy initially relied upon grouping taxa by morphological similarity. As the study of systematics became more and more sophisticated, new methods and characters, such as presence or absence of various chemicals or chemical pathways, were integrated into the view of how various groups and species of plants are related to one another.
Much more recently, the advent of DNA sequencing has revolutionized the field of systematics and how phylogenetic trees are constructed. This shift began in earnest in the mid-1990s, and one of the earliest genetic loci used in molecular phylogenetics of plants was rbcL, the gene that encodes the large subunit of the carbon-fixing protein Rubisco. As DNA sequencing technology has improved and become less and less expensive, computational speed and availability have allowed for large datasets of tens of thousands or even hundreds of thousands of molecular characters less prone to convergent evolution or incorrect interpretation of homology than morphological characters to be analyzed.
There are actually 3 different genomes within a plant cell: the nuclear genome where the majority of genes reside; the mitochondrial genome, which is a remnant of the bacterial genome that has become much reduced since the ancient endosymbiosis of an ATP-producing bacterium in the ancestor of all eukaryotes; and the chloroplast genome, the remnants of the cyanobacterial genome from the endosymbiotic event in the ancestor of Archaeplastida (Red Algae + Green Algae/Embryophytes). The nuclear genome is encoded on large, linear chromosomes while both mitochondrial and chloroplast genomes map to circular plasmids like their prokaryote precursors.
Despite its small size relative to the nuclear genome, the chloroplast genome has been the most widely-used genome for molecular systematics in plants to date. Chloroplast genes typically exist in single copy per genome, making homology easy to discern across all photosynthetic plants. Chloroplast genes also exhibit a slower rate of mutation than nuclear genes, allowing easy alignment of homologous characters from species to species. While rbcL, with ~1,500 alignable nucleotide characters, remains one of the most important loci used in plant systematics, modern sequencing technology and computational power has led to sequencing of the entire chloroplast genome for more and more species, which leads to datasets with 90,000+ aligned characters.
Mitochondrial genes are usually the slowest evolving genes in plants (by contrast, in animals they usually evolve faster than nuclear genes). While this makes them somewhat useful for inferring more ancient relationships, there are often not enough character changes for them to be informative below the family level. They are also prone to RNA editing (modification of some bases in the coding sequence after transcription), and, in some cases, horizontal gene transfer between unrelated plant species, both of which can mislead phylogeny. For parasitic plants that have lost the ability to photosynthesize and are missing or have highly modified chloroplast genes, mitochondrial genes are a necessary source for phylogenetic data. However, the problem of horizontal gene transfer from plant to plant is particularly widespread in cases of intimate host/parasite interactions.
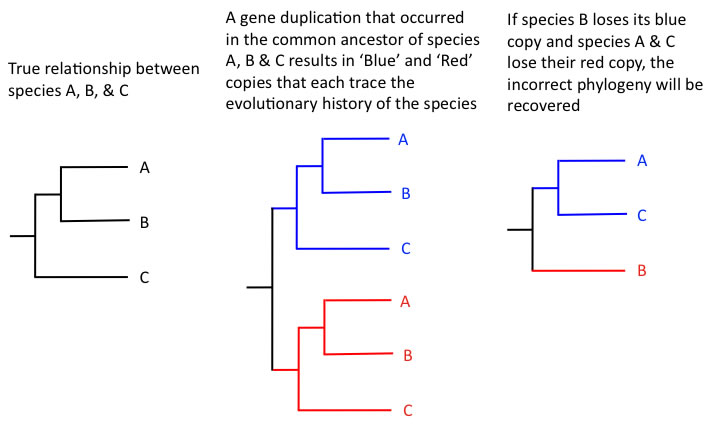
The nuclear genome contains many more genes than either organellar genome, but issues with paralogous gene copies make nuclear genes problematic enough that they have been underutilized as a source of phylogenetic data until recently. Paralogous genes are multiple copies of a gene retained after a gene duplication event. This can occur in many eukaryotes through segmental duplication- duplication of a section of a chromosome- or, particularly in plants, through polyploidy. Most lineages of plants have been through many polyploid events in their evolutionary history, leading to multiple divergent copies of most genes in the nuclear genome. For phylogenetic inference, it is imperative that the same copy of the gene is compared from species to species. In cases where one copy of a gene is deleted from one species, and the other copy is deleted from the other species, a strongly-supported, misleading phylogeny can be attained as diagrammed below.
Nuclear ribosomal RNA subunits (untranslated RNA copies that form the core of a ribosome) exist in high copy number on chromosomes, but the copies often recombine and 'fix' one another to make most copies identical. 3 of these RNA molecules are transcribed together, along with 2 'spacer' regions between them that must fold and be spliced out. These are known as the Internal Transcribed Spacer region (ITS), and although most noncoding regions evolve quickly, the portions involved in folding are more constained and help allow homologous bases to be aligned properly. Nonfolding regions evolve quickly enough to provide useful characters for species-level phylogeny. While ITS works well for some taxa, in other taxa it exists in multiple copies and behaves as poorly as other paralogous genes.
With more and more complete plant genomes as well as large datasets of transcribed genes from many other species, nuclear genes have become much more useful for phylogenetics in recent years. Many nuclear genes that are under strong selection to remain at single or low copy number and are less prone to problems with paralogy have been identified in recent years.
While molecular data have elucidated many aspects of plant phylogeny, it is important to note that many historical taxonomic groupings based on morphological similarity have been backed up by molecular data. Many families are easily recognizable thanks to morphological synapomorphies, characters shared by members of the family that evolved on the branch leading to their most recent common ancestor. These diagnostic groups are also supported by a large number of DNA synapomorphies as well.
In most cases where molecular phylogenies have changed traditional taxonomic groups, it is because a formerly recognized group has turned out to be paraphyletic. A paraphyletic group occurs when one or more species is not included in a group even though it is descended from the most recent common ancestor of that group. The example of "Lemnaceae," a diagnostic group of tiny floating aquatic plants traditionally recognized as their own family but now included within the family Araceae, is shown below.
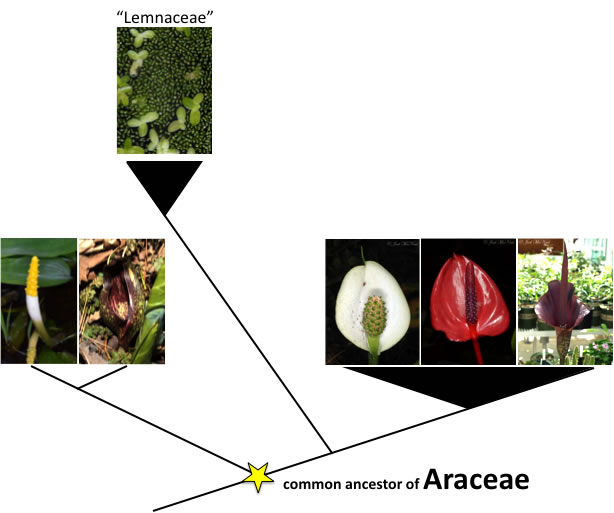
"Lemnaceae" are a monophyletic group that traces back to a shared common ancestor. Araceae, as traditionally recognized, were grouped based on a diagnostic spathe & spadix inflorescence. However, some Araceae are actually more closely related to "Lemnaceae" than they are to other Araceae. Although they are very tiny, the inflorescences of "Lemnaceae" are actually a submerged, microscopic spadix produced on the underside of the floating leaf and are descended from a common spathe & spadix-producing Araceae ancestor. "Lemnaceae" are really a lineage derived from within Araceae that have experienced extreme morphological changes due to their floating aquatic habit. To make Araceae monophyletic, "Lemnaceae" must be included in the family.
Molecular data are not immune to problems caused by a lineage changing drastically relative to more slowly evolving kin. When multiple lineages independently have accelerated rates of mutation, many characters may start to converge at rapidly evolving sites (particularly if those lineage share a common mutational bias, such as an increased likelihood of G/C mutations relative to A/T mutations). This phenomenon is known as long branch attraction. Methods of phylogenetic analysis that use models of sequence evolution to infer the correct tree, such as Maximum Likelihood, are not as easily mislead by these biases.




