


Research
My Research Interests
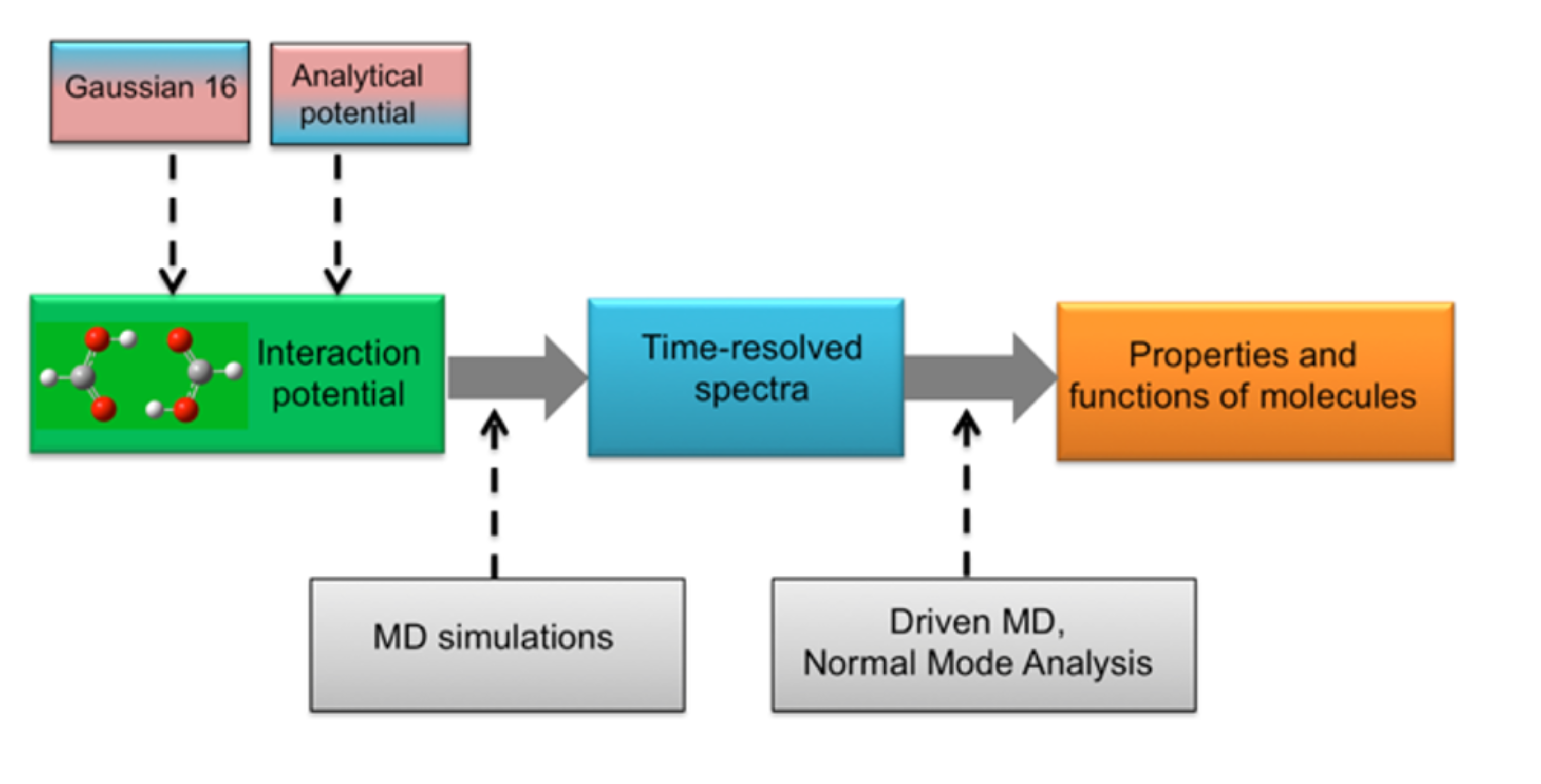
My research group uses a combination of theoretical and computational methods to understand and predict the behavior of molecules and chemical systems. We are particularly interested in the dynamics of hydrogen-bonded systems and carbon clusters at an atomic level. My goal is to help students gain hands-on experience, develop advanced skills, and contribute to the exciting field of computational chemistry.
• Computational Chemistry & Simulation: We use molecular dynamics simulators and various programming languages, such as Fortran,
C-shell, and Mathematica, to investigate chemical and physical properties of novel
materials and systems.
• Quantum Mechanics & Spectroscopy: Our work often involves studying the vibrational dynamics of molecules and clusters, including the use of molecular dynamics and driven molecular dynamics methods to simulate and assign spectra.
• Machine Learning: We explore the application of machine learning algorithms, such as those that use permutationally invariant polynomials, to fit potential energy surfaces and understand complex molecular systems.
• Computational Tools Development: A key part of our research is developing new computational tools and methods to study the structure and dynamics of molecular clusters more efficiently.
Why Should You Join My Research Group?
• Gain Hands-On Experience: Apply theoretical concepts to real-world computational problems.
• Enhance Your Skills: Develop proficiency in advanced programming, scientific computing, and data analysis.
• Publish Your Work: Contribute to scientific papers and present your findings at conferences.
• Prepare for the Future: Build a strong research portfolio for graduate school or a career in research and development.
How to Get Started?
• Review my recent publications posted on my website: https://facultyweb.kennesaw.edu/mkaledin/index.php
• Send me an email mkaledin@kennesaw.edu to set up a time to chat about your research interests.




