Variable Ethylene-Bridged Bis(amidines) and Bis(amidinates): Hydrogen Bonding and
Photoluminescence upon Deprotonation
Amidines are isolectronic N-analogs of carboxylic acids and esters that have attracted considerable attention
as building blocks for biomedical reagents, redox-switchable chromophores and fluorophores,
and also as versatile ligands in coordination chemistry.[1] Tethered bis(amidines) remained underrepresented in the literature until they were
rediscovered because of their striking similarity to bis(cyclopentadienyl) dianions
that play a key role as ligand scaffolds of lanthanide and Group IV ansa metallocenes,
which have revolutionized polymerization catalysis.[2] In particular, a series of alkylene- and arylene-linked bis(amidines) has been reported
to serve as convenient ligands for alkali metal, lanthanide, Group IV metal complexes,
and their related catalytic applications.[3] We have recently reported on a series of new flexible tetra- and hexadentate ethylene-linked
bis(amidines) that form versatile networks of inter- and intramolecular hydrogen bonds,
as revealed by X-ray crystallography and IR spectroscopy (Figs. 1–3).[4],[5]
Figure 2. XRD molecular structures of L1H2, L2H2, L6H2, and L8H2.
Figure 3. A supramolecular solvent adduct: L2H2·4EtOH.Combined experimental (1H NMR spectroscopy in different solvents) and computational studies (DFT, gas phase)
confirmed that hydrogen bonding for the less sterically crowded bis(amidine) L2H2 and all hexadentate congeners L5–9H2 is retained in solution. We also discovered that L1–9H2 become substantially emissive upon deprotonation through nBuLi (or NaN(SiMe3)2). Thus, L1–9H2 and their dianions represent a series of unprecedented photoluminescent bis(amidine)/bis(amidinate)
light switches.
Scheme 1. Deprotonation of bis(amidines); THF solution of [L5Li2] under exposure of UV light; steady-state emission spectra.
References
Novel Multinuclear Coinage Metal Cluster Arrays to Design Molecular Light-Emitting
Devices
Linear multi-atom complexes of Group 11 metals (Cu, Ag, Au) exhibit fascinating luminescence
properties that make them highly interesting
1) for the systematic design of molecular wires in nanoelectronics
and
2) their application as light-emitting building blocks in organic light-emitting devices/diodes
(OLEDs)[1–3].
Since the light-emitting properties are specifically dependent on the number of metal
atoms, these complexes can directly be applied in new tailored OLEDs, particularly
in those that emit blue light – an urgent interest in the development of next-generation,
energy-saving light sources.[1],[2],[4] Previous methods to synthesize multi-atom complexes have shown that these compounds
are usually available in only low yields, as contaminated product mixtures, or they
are not accessible at all.
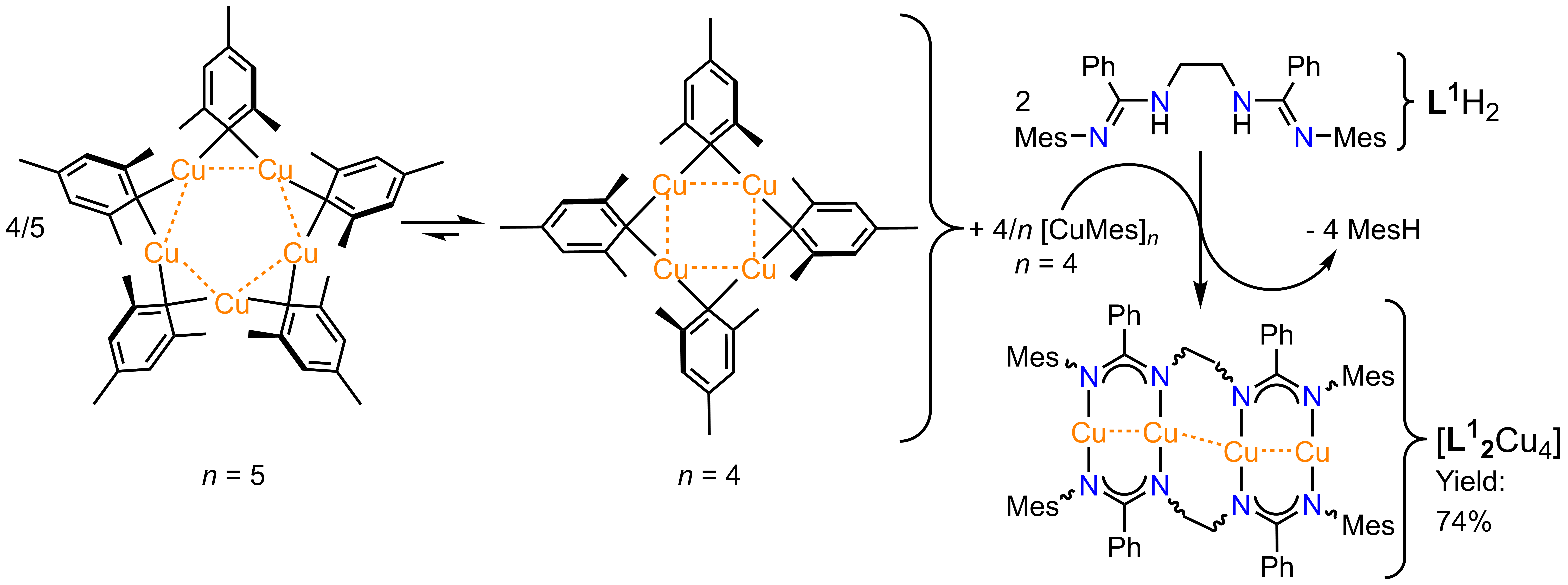
We have utilized a new synthetic method to incorporate CuI ions into a larger bis(amidinate) ligand framework [L1]²¯ under ambient conditions and with excellent yields (Scheme 1).[5]
The star-shaped organometallic compound mesitylcopper, [CuMes]n (n = 4, 5),[6] is a clean source of copper ions, since it serves as deprotonating base itself and
produces only one volatile organic byproduct (mesitylene, MesH).
Scheme 1. Clean synthesis of [L12Cu4] from mesitylcopper.
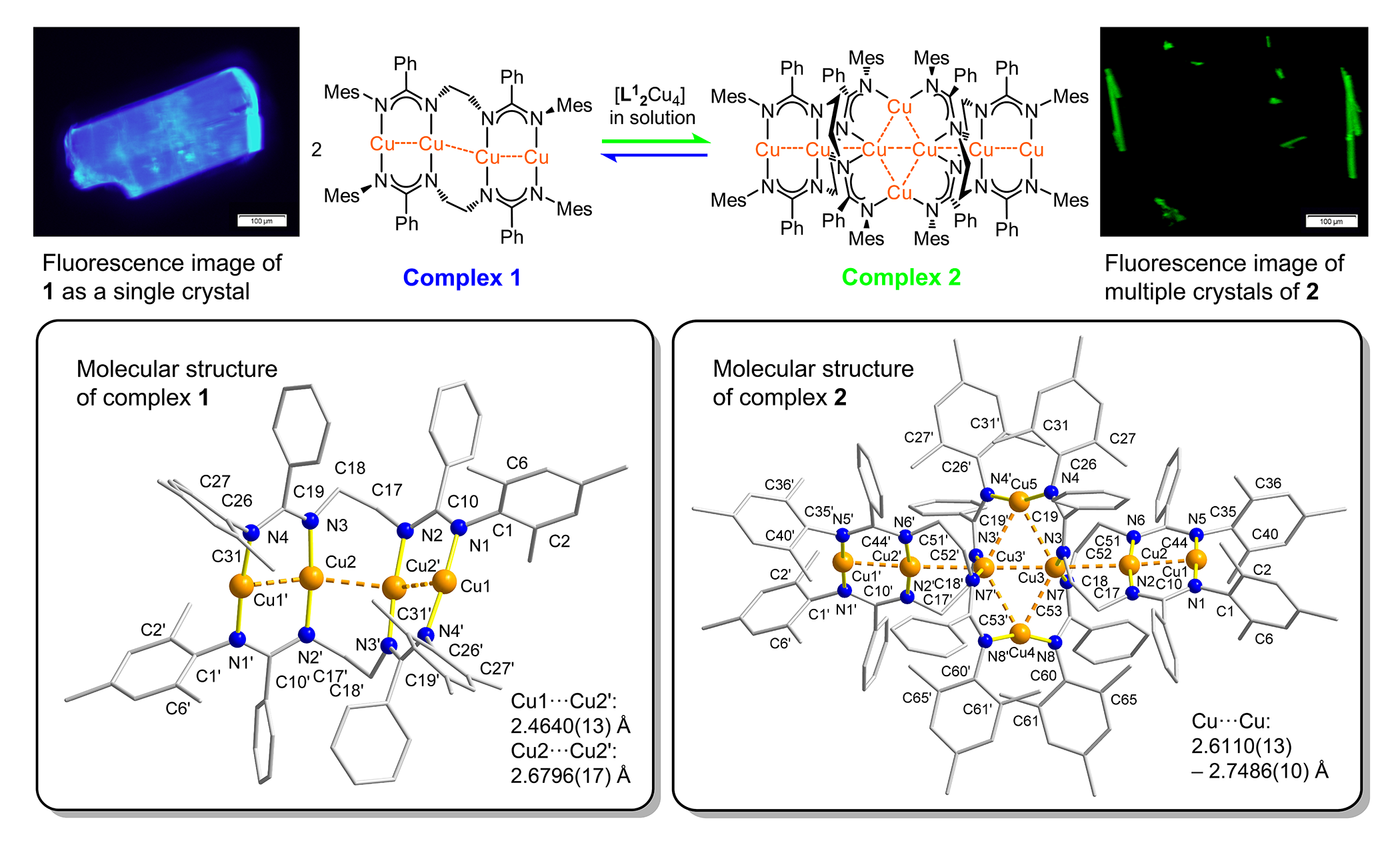
This new method allows us to control the size of (strictly linear or helically-bent)
copper chain complexes: If [L12Cu4] crystallizes from solution, it forms two different complexes 1 and 2 (Scheme 2), depending on how polar the solvent mixture is. Toluene/hexanes mixtures
yield both crystals of 1 and 2 simultaneously while more polar toluene/diethyl ether mixtures result in the exclusive
formation of 2. As revealed by X-ray crystallography, a method to determine a molecular structure
in the solid state, our new ligand [L1]²¯ accommodates four copper atoms in a linear, coiling-like arrangement (1).
Complex 1 can adopt a higher-level organization that creates an even longer chain of six copper
atoms with two additional bridging central Cu(I) centers (2). This is one of the longest linear arrangement of Cu(I) ions in a discrete molecule
reported so far. An important criterion for a molecular wire are the distances between
the metal atoms: If they are, in case of copper, in the magnitude of –or smaller than–
2.8 Å (the sum of two copper van der Waals radii), then they undergo significant contact
interactions.
Scheme 2. Formation of complexes 1 and 2 as crystalline solids from solution. Fluorescence images of crystals of 1 and 2. Molecular structures of 1 and 2, determined by X-ray crystallography.
1 and 2 can directly be applied in OLEDs, since the two distinct crystal morphologies show
blue (λmax = 460 nm; 1) and green (λmax = 495 nm; 2) light emissions (Scheme 2) in excellent singlet-to-triplet interconversions (a measure
how efficient an emissive material is) – these are outstanding features for direct
applications in OLEDs. Supported by computational calculations, we could demonstrate
that the increased number of contacts between copper atoms in 2 relative to 1 results in the observed longer emission wavelength.
We have recently reported on a straight tetracopper array with a centrally disconnected
CuI2···CuI2 chain and another helically bent CuI4 complex that forms a unique bis(amidinate) cluster comprising ten CuI ions.[7] The tetranuclear complexes exhibit blue phosphorescence or TADF (thermally activated
delayed fluorescence), which is dependent from the available void space in their crystal
lattices. Additional N-donor groups in the bis(amidinate) moieties result in a new class of unusual intertwined
arrangements.[8]
Overall, we have applied an efficient method to synthesize unique, highly luminescent
multi-atom copper complexes that represent linear and coiling-like molecular wires.
These compounds are excellent candidates for applications in OLEDs and prospective
multifunctional nanoelectronic devices. Our current research projects are also concerned
with new bis(amidinate) complexes of the heavier coinage metals (Ag and Au).
References
[1] a) Yersin, H., Ed. Highly Efficient OLEDs with Phosphorescent Materials; Wiley-VCH: Weinheim, Germany, 2008. b) Yersin, H.; Rausch, A. F.; Czerwieniec,
R.; Hofbeck, T.; Fischer, T. Coord. Chem. Rev.2011, 255, 2622–2652. [2] Stollenz, M. Chem. Eur. J.2019, 25, 4274–4298. [3] Yam, V. W.-W.; Au, V. K.-M.; Leung, S. Y.-L. Chem. Rev.2015, 115, 7589–7728. [4] Sasabe, H.; Kido, J. Eur. J. Org. Chem.2013, 7653–7663. [5] Stollenz, M.; Raymond, J. E.; Pérez, L. M.; Wiederkehr, J.; Bhuvanesh, N. Chem. Eur. J.2016, 22,
2396–2405. [6] Stollenz, M.; Meyer, F. Organometallics2012, 31, 7708–7727.
[7] Arras, J.; Calderón-Diaz, A.; Lebedkin, S.; Gozem, S.; McMillen, C. D.; Bhuvanesh,
N.; Stollenz, M. Inorg. Chem.2024, 63, 12943–12957.
[8] Dowling, K.; Kruczyński, T.; Dutta, S.; Ngan Le, D. P.; Gozem, S.; McMillen, C.
D.; Stollenz, M. Chem. Commun.2024, 60, 11136–11139.
N−H···Cl−M and N−H···C−M Hydrogen Bonds in Group 11 Metal Coordination Spheres
This project addresses fundamental questions on how the combination of weak intramolecular
forces stabilizes energetically preferred conformations of coordination compounds
and organometallic complexes, which is relevant to supramolecular architectures, reaction
mechanisms, and catalysis.
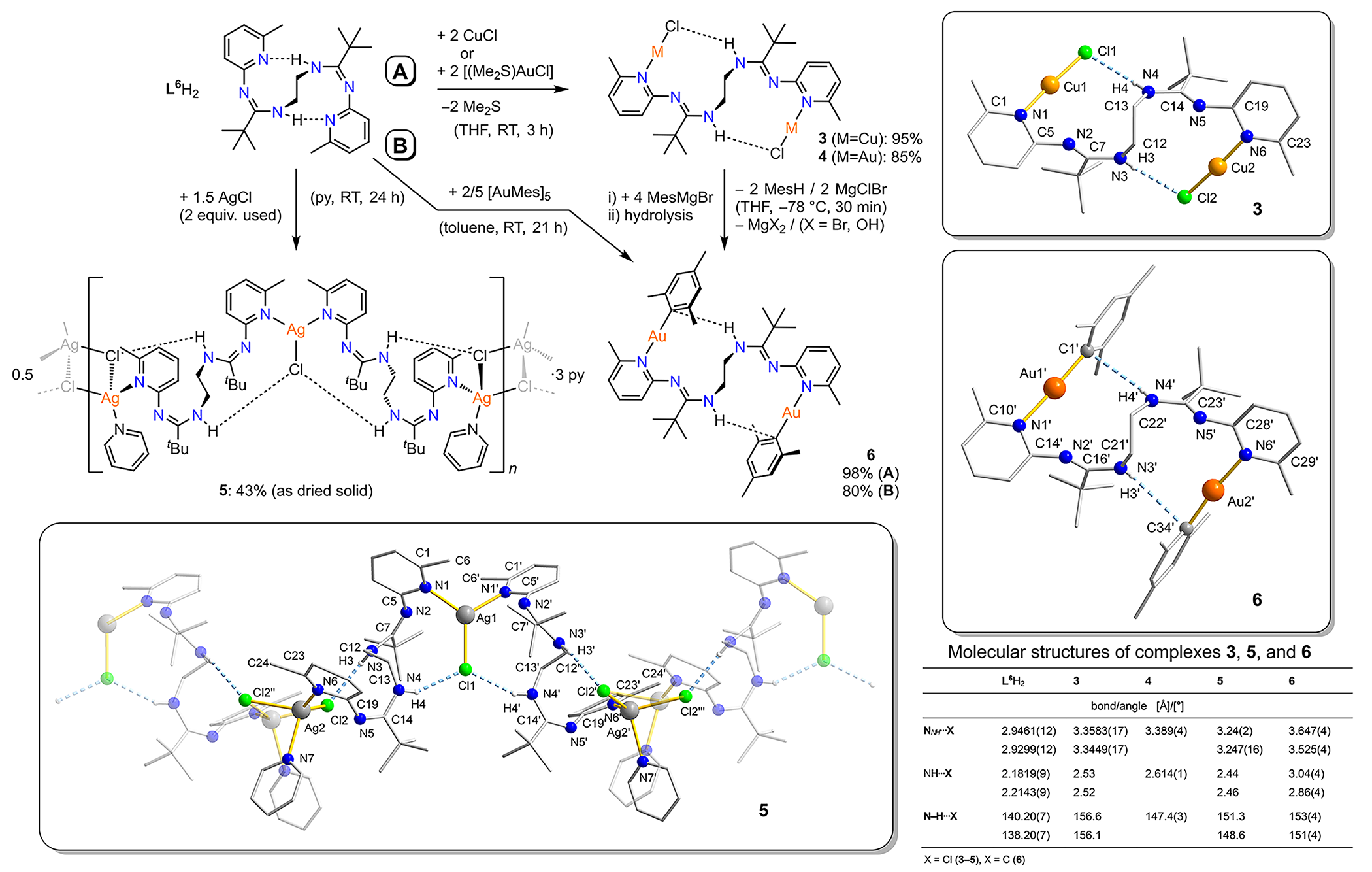
Bis(amidine) ligand L6H2 is capable of incorporating distinct CuCl, AuCl, or even AuMes complex fragments
(Mes = mesityl) of the pentameric cluster [AuMes]5[1] through formal insertions into the two intramolecular NH···N' hydrogen bonds of its
pyridyl/amidine binding pockets (Scheme 3).[2],[3] X-ray crystallography revealed that the resulting dicopper and digold complexes 3,4, and 6 show NH···acceptor–M (M = Cu, Au) hydrogen bonding, are isostructural, exhibit C2 symmetry, and are chiral. Their crystal packings contain racemic mixtures of both
enantiomers. Complex 6 features two rare non-conventional NH···Cipso–Au hydrogen bonds that represent primal onsets of proton transfer reactions in an
organometallic complex. Quantum Theory of Atoms in Molecules (QTAIM) calculations[4] on 3, 4, and 6 support the existence of additional intramolecular London dispersion forces, which
altogether contribute to the observed conformations of the binuclear complexes.
Scheme 3. Synthesis of complexes 3−6 and molecular structures of 3, 5, and 6, determined by X-ray crystallography.
Several efforts to obtain an isostructural complex [L6H2(AgCl)2] were unsuccessful which is attributed to the poor solubility of AgCl in most common
organic solvents. However, pyridine was found convenient to produce a homogeneous
solution of L6H2 and AgCl that formed the coordination polymer {(L6H2)2(py)2(AgCl)3](py)3}n (5, Scheme 2). X-ray crystallography revealed the presence of trigonal-planar and
tetrahedral coordination geometries of two distinct sorts of Ag+ ions in the polymeric chain.
Variable-temperature 1H NMR spectra of 3, 4, and 6 (CDCl3 and C6D6) show essentially no change across a wide temperature range. However, there is decoalescence
of the broad CH2 resonance signal into two singlets originating from diastereotopic CH2 protons (Scheme 4). Disruption of the hydrogen bonds would be expected to result
in an upfield-shift of the N–H signals, which is not observed. For the solution state,
this suggests a concerted conformational inversion of the double ring system that
retains the two N–H···acceptor hydrogen bonds. As a result, a reversible interconversion
from one C2-enantiomer into the other through the formation of a transient Ci-symmetrical intermediate is observed. The subtle interplay of molecular design, hydrogen
bonding, and London dispersion forces leads to fundamental insights in controlling
supramolecular assemblies and new reactivity.
Scheme 4. Variable-temperature 1H NMR spectra of complex 3, free energies of activation for 3, 4, and 6, computational structure of the Ci-symmetrical intermediate of 3.
References
[1] a) Gambarotta, S.; Floriani, C.; Chiesi-Villa, A.; Guastini, C. A. J. Chem. Soc., Chem. Commun.1983, 1304–1306. b) Meyer, E. M.; Gambarotta, S.; Floriani, C.; Chiesi-Villa, A.; Guastini,
C. A. Organometallics1989, 8, 1067–1079. c) Usón, R.; Laguna, A.; Fernández, E. J.; Ruiz-Romero, M. E.; Jones,
P. G.; Lautner, J. J. Chem. Soc., Dalton Trans.1989, 2127–2131. [2] Arras, J.; Ugarte Trejo, O.; Bhuvanesh, N.; Stollenz, M. Chem. Commun.2022, 58, 1418–1421. [3] Arras, J.; Ugarte Trejo, O.; Bhuvanesh, N.; McMillen, C. D.; Stollenz, M. Inorg. Chem. Front.2022, 9, 3267–3281. [4] Bader, R. F. W. Chem. Rev.1991, 91, 893–928.



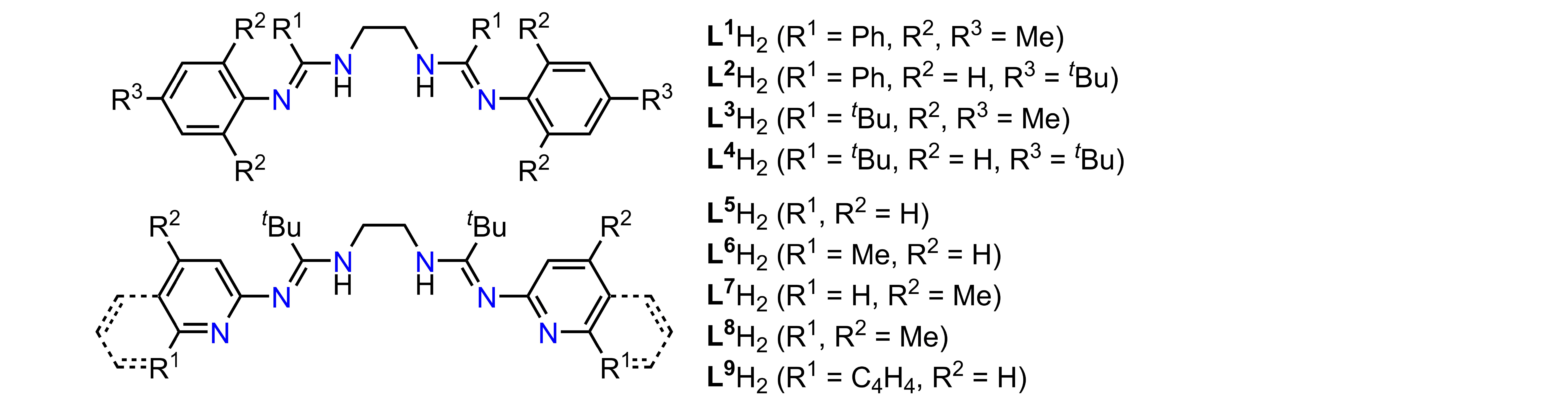 Figure 1. Ethylene-bridged N,N'-disubstituted bis(amidines) L1–9H2.
Figure 1. Ethylene-bridged N,N'-disubstituted bis(amidines) L1–9H2.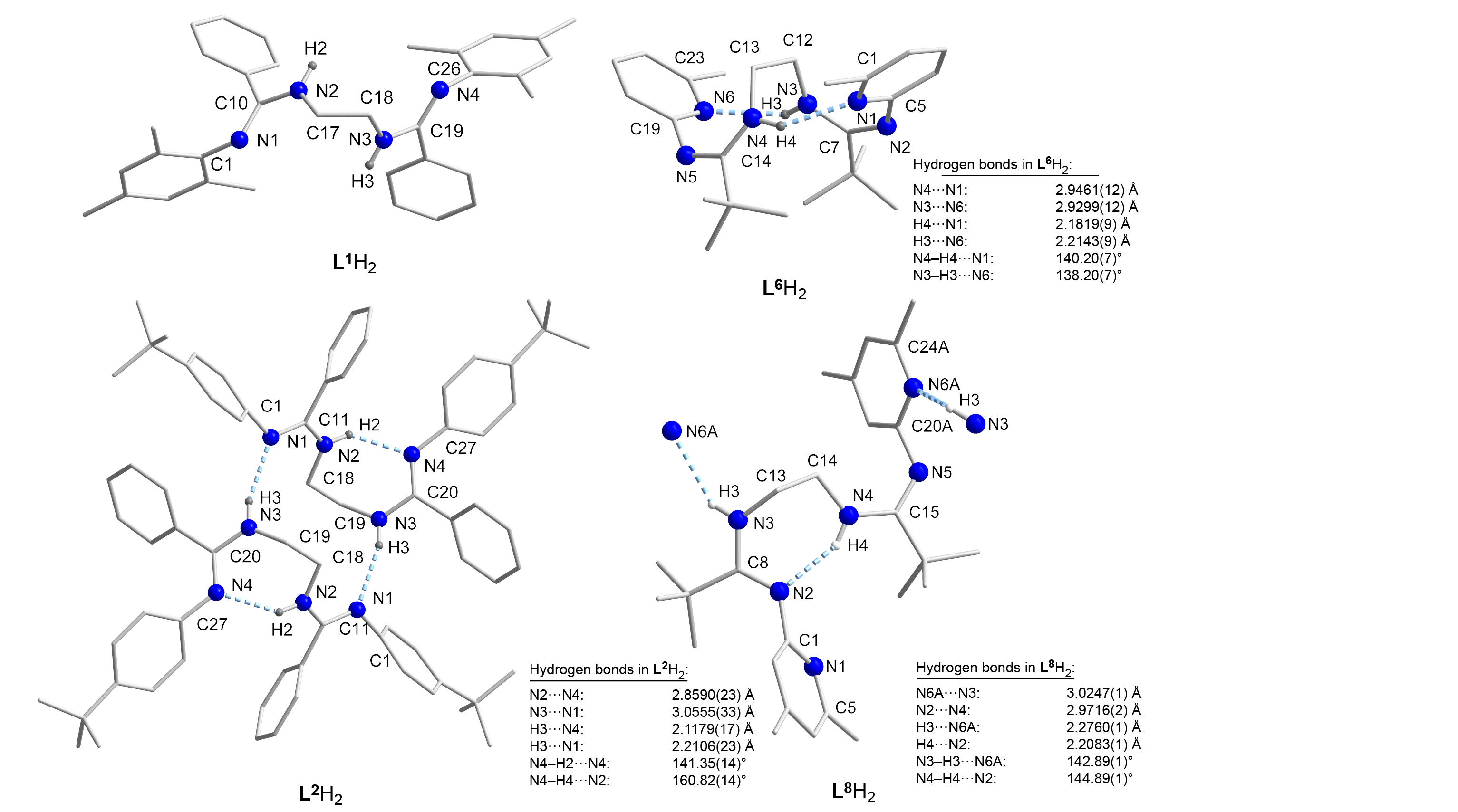
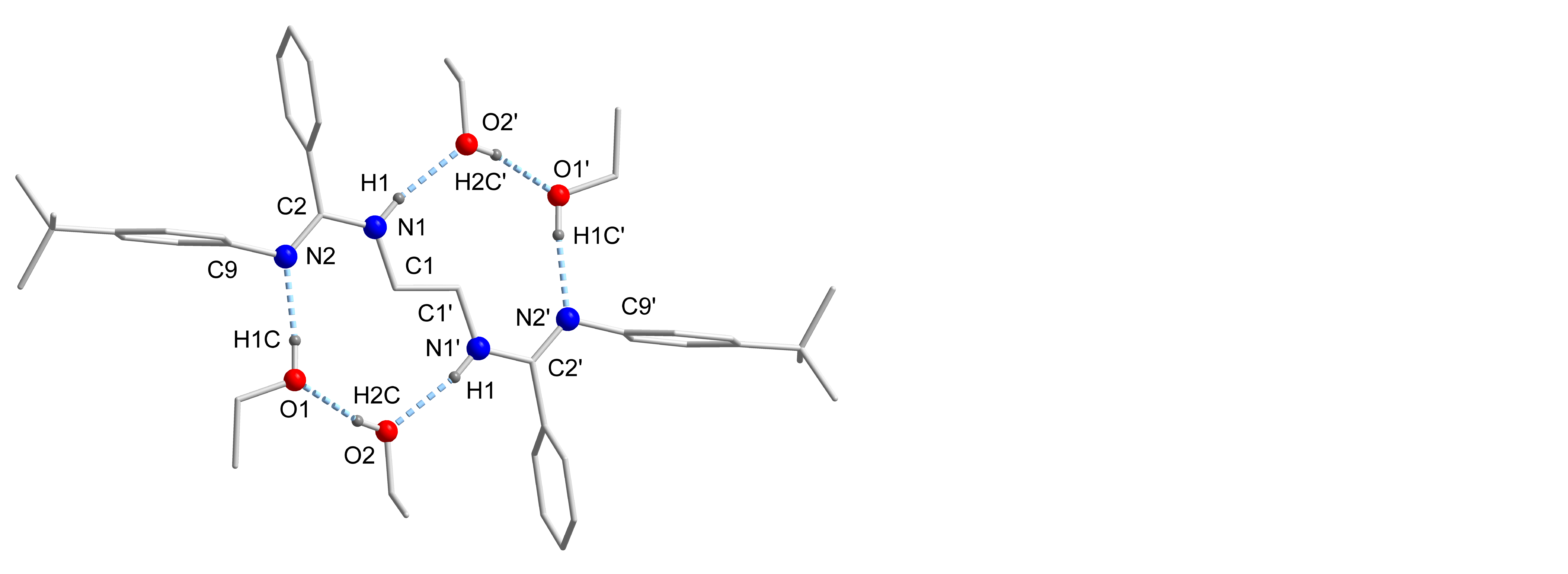 Figure 3. A supramolecular solvent adduct: L2H2·4EtOH.Combined experimental (1H NMR spectroscopy in different solvents) and computational studies (DFT, gas phase)
confirmed that hydrogen bonding for the less sterically crowded bis(amidine) L2H2 and all hexadentate congeners L5–9H2 is retained in solution. We also discovered that L1–9H2 become substantially emissive upon deprotonation through nBuLi (or NaN(SiMe3)2). Thus, L1–9H2 and their dianions represent a series of unprecedented photoluminescent bis(amidine)/bis(amidinate)
light switches.
Figure 3. A supramolecular solvent adduct: L2H2·4EtOH.Combined experimental (1H NMR spectroscopy in different solvents) and computational studies (DFT, gas phase)
confirmed that hydrogen bonding for the less sterically crowded bis(amidine) L2H2 and all hexadentate congeners L5–9H2 is retained in solution. We also discovered that L1–9H2 become substantially emissive upon deprotonation through nBuLi (or NaN(SiMe3)2). Thus, L1–9H2 and their dianions represent a series of unprecedented photoluminescent bis(amidine)/bis(amidinate)
light switches.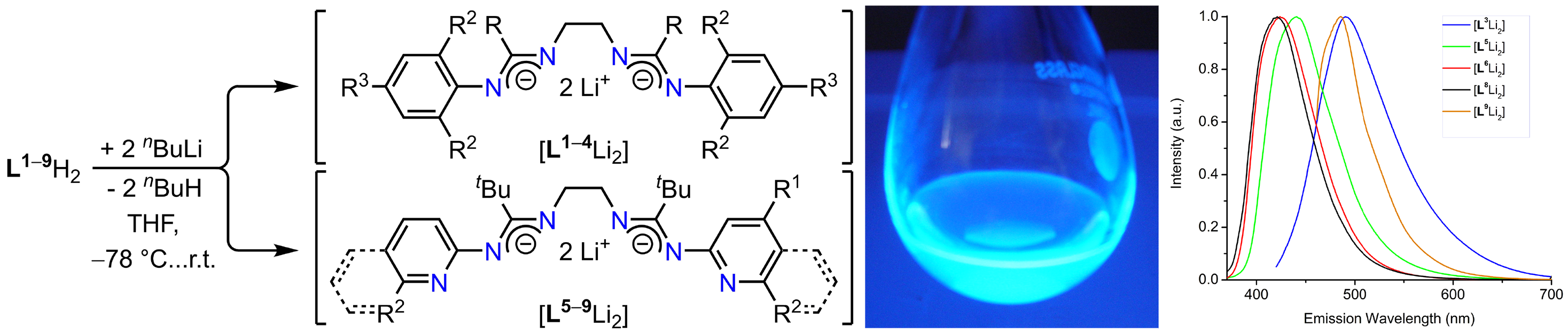
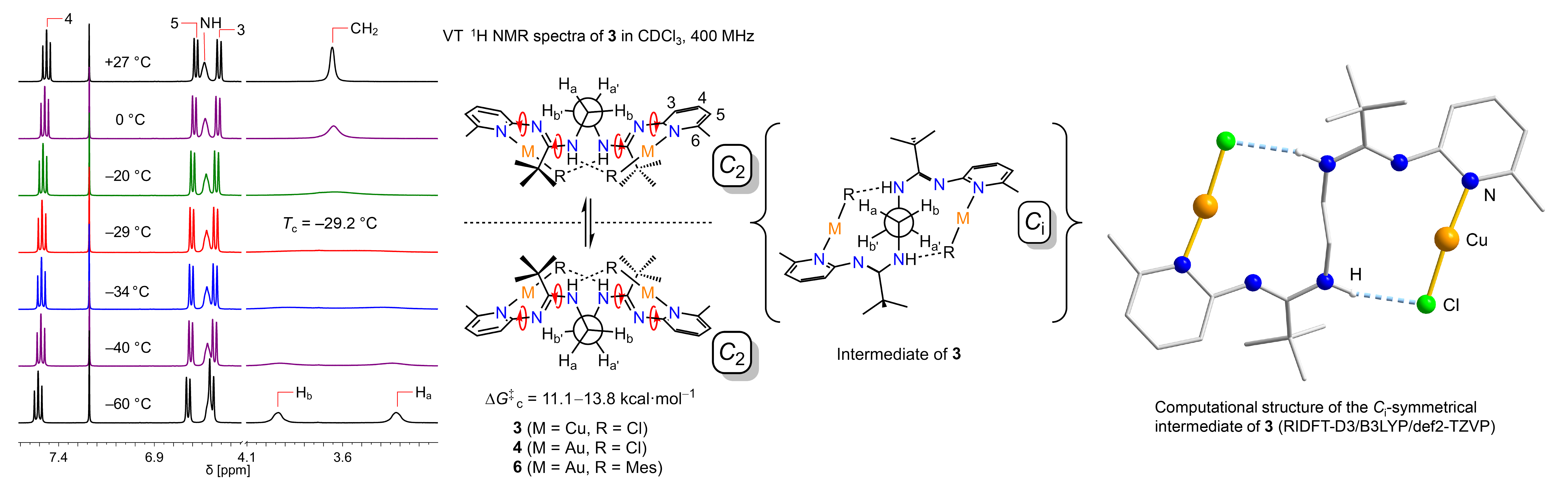 Scheme 4. Variable-temperature 1H NMR spectra of complex 3, free energies of activation for 3, 4, and 6, computational structure of the Ci-symmetrical intermediate of 3.
Scheme 4. Variable-temperature 1H NMR spectra of complex 3, free energies of activation for 3, 4, and 6, computational structure of the Ci-symmetrical intermediate of 3.



